VIII. Определение молекулярного профиля бруцелл с использованием полногеномного секвенирования
8.1. Максимальную разрешающую способность для генетического типирования бруцелл демонстрирует выявление стабильных одиночных нуклеотидных полиморфизмов при анализе полногеномных последовательностей.
Методика заключается в оценке вариабельности полиморфизмов в локусах с высокой степенью полиморфизма, на основании которой определяются филогенетические взаимосвязи изолятов бруцелл.
8.2. Концентрацию образцов ДНК определяют на флуориметре, в работе используют пробы, содержащие не меньше 500 нг ДНК. Оценку чистоты образцов проводят путем определения отношения A260/280 с использованием спектрофотометра. Значение соотношения A260/280 для чистых образцов ДНК должно находиться в диапазоне 1,8 - 2,0.
8.3. Фрагментацию геномной ДНК бруцелл до длины ~ 400 п.н. проводят физическим методом в системе ультразвуковой обработки. Можно проводить фрагментацию ДНК до требуемой длины с использованием набора реагентов, содержащего ферменты рестрикции.
Контроль длины фрагментов библиотек осуществляют с помощью микрокапиллярного электрофореза, концентрацию ДНК-библиотек определяют с помощью флуориметра.
К полученным фрагментам ДНК путем лигирования присоединяют универсальные олигонуклеотидные адаптеры с известной последовательностью нуклеотидов. Эти адаптеры необходимы для последующей амплификации (гибридизации) библиотеки фрагментов. Все этапы подготовки и обогащения библиотек ДНК проводят по стандартным протоколам производителей наборов реагентов.
8.4. Фрагменты ДНК-библиотеки гибридизируют с адаптерами, иммобилизованными на поверхности микросфер, с использованием системы эмульсионной ПЦР. Процедуру обогащения микросфер выполняют с помощью системы очистки и магнитных частиц. Каждый этап проводят по стандартным протоколам производителя наборов реагентов.
8.5. На этапе секвенирования микросферы, содержащие амплифицированные фрагменты ДНК-библиотек, помещают на чип. Секвенирование проводится путем синтеза комплементарной цепи с детекцией изменения значения pH в лунках чипа, возникающего при присоединении нуклеотида. Изменение pH считывается прибором и преобразуется встроенным программным обеспечением в цифровой сигнал, который в свою очередь преобразуется в буквенную последовательность нуклеотидов.
Все манипуляции выполняются в соответствии со стандартными протоколами.
8.6. Для оценки качества данных секвенирования используют показатель качества "Phred" (Q), который логарифмически связан с вероятностью ошибочного определения нуклеотида (P) и рассчитывается по формуле (2):
Значение показателя качества Q10 соответствует вероятности некорректного определения нуклеотида как 1 из 10 (степень достоверности 90,0%), Q20 как 1 из 100 (степень достоверности 99,0%), Q30 как 1 из 1000 (степень достоверности 99,9%) и т.д. Значение показателя качества Q20 является допустимым или пороговым уровнем для определения достоверности результатов. Каждому нуклеотиду в последовательности соответствует определенный показатель качества, т.е. степень достоверности определения нуклеотида (рис. 16).
На графике, представленном на рис. 16, по оси X откладывается позиция нуклеотида в риде, по оси Y - его качество. Область на графике, окрашенная в зеленый цвет, включает нуклеотиды с высоким качеством (показатель Q от 28 до 50), желтая - с допустимым (показатель Q от 20 до 28), красная область - нежелательным (показатель Q от 0 до 20). Горизонтальная синяя линия показывает среднее значение качества, красная линия внутри каждого желтого блока - медиану.
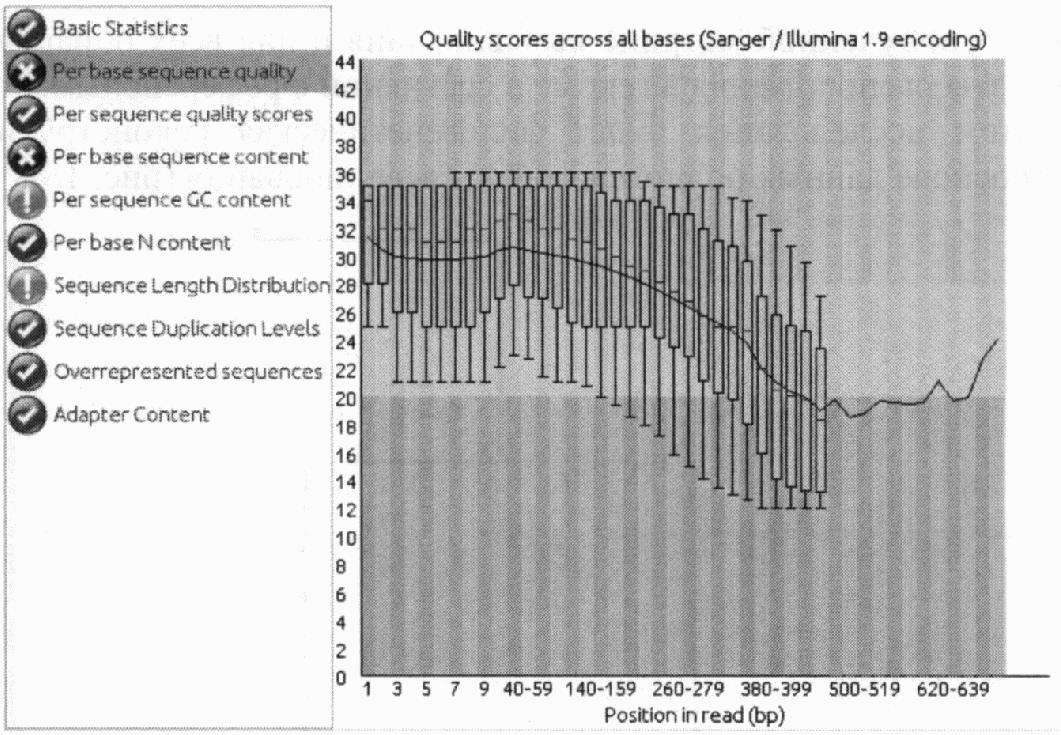
Рис. 16. Гистограмма оценки качества данных секвенирования
штамма B. melitensis И-349
8.7. Низкокачественные риды со средним значением баллов качества Q < 20, а также риды длиной менее 35 нуклеотидов удаляют (рис. 17).

Рис. 17. Гистограмма оценки качества данных секвенирования
штамма B. melitensis И-349 после фильтрации
8.8. Сборка генома осуществляется методом картирования на референсный геном. Для работы с геномной последовательностью используют контиги длиннее 500 нуклеотидов.
8.9. Коровый геном штаммов бруцелл получают путем множественного выравнивания изучаемых полногеномных последовательностей против референсного генома штамма соответствующего вида рода Brucella.
Множественное выравнивание выполняют с помощью программного обеспечения, которое позволяет получить матрицу множественного выравнивания геномов, содержащую только гомологичные нуклеотидные последовательности, все паралогичные последовательности исключают из анализа. Проводят поиск единичных нуклеотидных полиморфизмов в полученной матрице выравнивания и для всех обнаруженных определяют координаты расположения в геноме в сравнении с референсным.
Результатом исследования будет филогенетическая реконструкция штаммов Brucella spp. на основе данных полногеномного секвенирования (рис. 18).
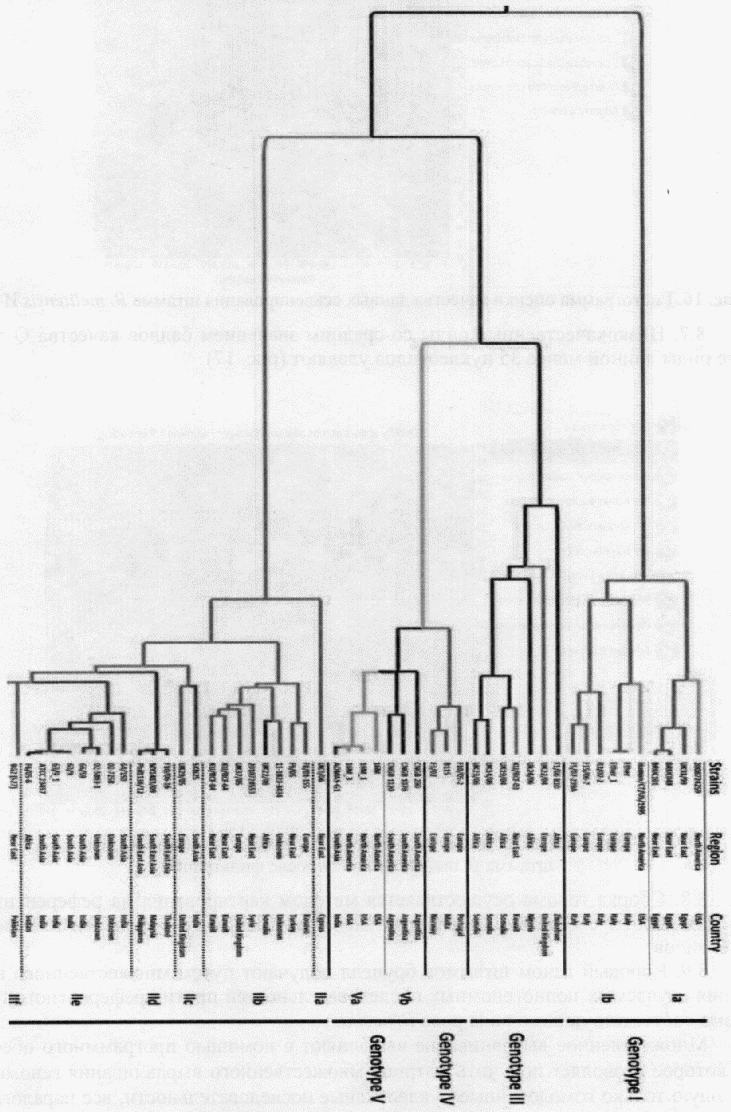
Рис. 18. Филогенетическое дерево штаммов B. melitensis,
построенное по результатам полногеномного анализа
- Гражданский кодекс (ГК РФ)
- Жилищный кодекс (ЖК РФ)
- Налоговый кодекс (НК РФ)
- Трудовой кодекс (ТК РФ)
- Уголовный кодекс (УК РФ)
- Бюджетный кодекс (БК РФ)
- Арбитражный процессуальный кодекс
- Конституция РФ
- Земельный кодекс (ЗК РФ)
- Лесной кодекс (ЛК РФ)
- Семейный кодекс (СК РФ)
- Уголовно-исполнительный кодекс
- Уголовно-процессуальный кодекс
- Производственный календарь на 2025 год
- МРОТ 2026
- ФЗ «О банкротстве»
- О защите прав потребителей (ЗОЗПП)
- Об исполнительном производстве
- О персональных данных
- О налогах на имущество физических лиц
- О средствах массовой информации
- Производственный календарь на 2026 год
- Федеральный закон "О полиции" N 3-ФЗ
- Расходы организации ПБУ 10/99
- Минимальный размер оплаты труда (МРОТ)
- Календарь бухгалтера на 2026 год
- Частичная мобилизация: обзор новостей
- Постановление Правительства РФ N 1875